
Surgical Implications
Burke Robinson, MD, Peter Hilger, MD
In the evaluation of a patient for cosmetic surgery, an underlying congenital origin for the deformities noted, with possible expression in multiple organ systems, should always be considered. In this case, a 19 year old woman requesting rhinoplasty exhibited facial anomalies: lack of quadrangular and lower lateral cartilages, small bony dorsum, underdeveloped aural cartilage, and infantile larynx and trachea. Otherwise healthy, she recalled having recurrent bouts of croup until she was 15 years old, exercise limitation secondary to stridor, and a family history of nasal hypoplasia. Genetic referral diagnosed her condition as Optiz-BBB/G compound syndrome (oculogenitolaryngeal syndrome) and we here review its head and neck manifestations. Suspected agenesis of nasal cartilage, unreported in previous literature (to our knowledge), was confirmed intraoperatively and presented a unique challenge for reconstruction, including a restricted choice of anesthesia and a need for banked cartilage. (Arch Otolaryngol Head Neck Surg. 1989; 115:985-988)
During a consultation for cosmetic surgery, the facial plastic surgeon should consider and underlying congenital cause that might account for the deformities noted. These deformities may be part of a syndrome that is expressed and identifiable in multiple organ system. The preoperative diagnosis of such a disorder adds information about any underlying skeletal abnormalities allowing for completed and safe surgical planning that can help to avoid intraoperative pitfalls. We present the case of a young woman desiring rhinoplasty who was found to have Optiz-BBB/G compound syndrome. Her abnormalities adversely affected both her larynx and trachea, and caused complete agenesis of all nasal cartilages. As a result of taking these into account in preoperative planning, an anesthetic complication was avoided, multiple tissue sources were harvested for her nasal reconstruction, and she, along with her family, received through genetic counseling.
REPORT OF A CASE
A 19 year old woman sought consultation for a cosmetic rhinoplasty. Physical examination demonstrated absence of the quadrangular septal cartilage, a retracted columella, and a short bony dorsum with low projection. There were fleshy alar lobules with lower lateral cartilages that were either very flaccid or absent. The nasal tip was over-rotated and lacked definition and projection. There were creases in the region where the soft triangle is normally located (Figs 1 and 2).
In addition the patient had hypertelorism exaggerated by her nasal deformity (Fig 3). The vertical dimension of her upper lip was increased, relative to the projection of her nose. Finally, her ears had remarkably small, flaccid auricular cartilages. Her mother (also present at the consultation) had similar nasal hypoplasia and anomalies of the ears.
Additional questioning heightened the level of suspicion for an underlying congenital origin of these anomalies. The patient’s medical history revealed “recurrent croup.” Further investigation showed that at the age of 16 years, the patient had required emergency intubation and her trachea had barely admitted a No. 4 endotracheal tube: the endoscopic report revealed an “infantile trachea.” At the age of 19 years, she was still experiencing intermittent episodes of stridor and, with exertion; dyspnea. Indirect laryngoscopy revealed a relatively small larynx and an upper trachea narrowed laterally.
The patient was referred for genetic workup to the University of Minnesota, Minneapolis, where a rare hereditary disorder, the Optiz-BBB/G compound syndrome, was diagnosed. The classic phenotypic expression shows hypertelorism, hypospadias, and laryngotracheoesophageal defects (Fig 4). Hypoplasia of the vocal cords, epiglottis, or the entire larynx has been reported, as well as absence of the dorsal portions on the larynx and trachea. The bony nasal dorsum in Optiz-BBB/G has been described as elevated or as flattened with anteverted nostrils and a rather flat, inapparent philtrum. The present case adds another observed expression to the phenotype, agenesis of nasal cartilage, and an explanation of some of the previously described nasal deformities.
RESULTS
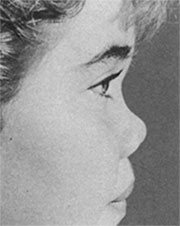
Fig 1. – Preoperative nasal presentation (lateral view)
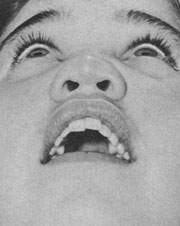
Fig 2. – Preoperative nasal presentation (base view))

Fig 3. – Preoperative nasal presentation (anterior view)
Local anesthesia with sedation was chosen for the surgical procedure because of the abnormal airway that would not guarantee safe intubation for general anesthesia. A composite graft of conchal cartilage with overlying skin measuring 1x 4 cm was harvested (Fig 5). Standard external rhinoplastic incisions were made. Elevation of nasal skin revealed myxoid fibrous tissue where the lower lateral cartilages should be (Fig 6), and histologic examination later demonstrated unorganized smooth muscle, fibrous tissue, and nerves (Fig 7). No septal or upper lateral cartilages were found.
The nasal mucosa was incised at the inferior border of the nasal bones. This allowed half of the nose to be repositioned more inferiorly and resulted in lengthening of the nasal dorsum. The auricular composite graft was placed into the newly created defect at the inferior edge of the nasal bones, with the cutaneous surface placed intranasally, and this was sutured into place with 4-0 polyglactin 910 (Vicryl) sutures ( Figs 6 and 8 ). Next irradiated homograft cartilage available because we had anticipated the need, based on the patient’s physical examination and genetic evaluation was carved into a columellar strut with a dowel on one end. Another piece of cartilage was shaped to provide a dorsonasal support with an acetabulum. This was secured to the root of the nose with a 9 mm titanium selftapping screw inserted through a stab wound at the root of the nose (Fig 9). The columellar strut was positioned in the membranous septum and its dowel interdigitated with the acetabulum of the dorsal graft (Fig 10). And additional piece of cartilage was sculptured in the form of an extended Sheen graft and secured too the columellar and dorsal struts with 4-0 polyglactin 910 sutures to improve projection and definition of the nasal tip (Figs 11 and 12). The nasal skin was redraped and sutured with 4-0 and 6-0 chromic sutures.
The postoperative photographs demonstrate improvement in nasal projection and length as well as projection and definition of the tip (Fig 13). A columellar composite graft is planned to improve columellar show and to provide additional projection of the tip. Genetic counseling was provided to the patient and her family concerning probability of transmission of the condition. If she should have sons, for example, each son would have a 50% chance of being born with hypospadias. The patient was also advised that if future offspring exhibit laryngoesophageal insufficiency and/ or nasolaryngeal hupoplasia, surgical intervention could be performed based on adequate knowledge of the syndrome.
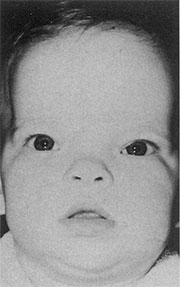
Fig 4. – Classic facies in the Optiz-BBB/G syndrome.
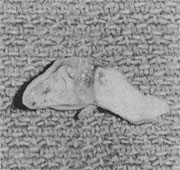
Fig 5. – Intraoperative photograph showing composite auricular graft.
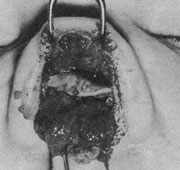
Fig 6. – Intraoperative photograph demonstrating mixoid fibrous tissue and placement of composite auricular graft.
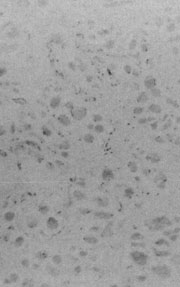
Fig. 7. – Histologic examination of mixoid fibrous tissue revealed unorganized smooth muscle, fibrous tissue, and nerves (hematoxy-lin-eosin, X 100).
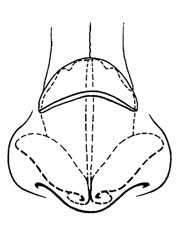
Fig. 8. – Artist’s sketch depicting placement of auricular graft.
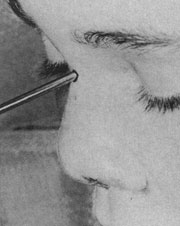
Fig. 9. – Intraoperative photograph showing homograft nasal support, secured with titanium screw.
COMMENT
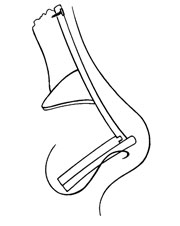
Fig. 10. – Artist’s sketch (lateral view)
The Optiz-BBB/G compound syndrome was originally described by Optiz as two separate genetic syndromes. The BBB syndrome consisted of hypertelorism, hypospadias, and other subtle anomalies, while the G syndrome added laryngotracheoesophageal defects. Subsequent reports of BBB, however, implied that laryngeal problems were also involved (eg, “suffocation” at 8 weeks of age and laryngeal stridor in a 3 year old) and documented common clinical manifestations, a similar autosomal dominant inheritance pattern, and increased severity of expression on males. The two syndromes have recently been discussed in combination, either as the same condition or as allelism.
Manifestationsin in the head and neck are many and vary in the degree of expression. Hypertelorism in common and may be the only expression in females. In addition, upslanting or downslanting palpebral fissures, strabismus, and epicanthal folds have been reported. Pinnae may demonstrate an abnormal helix and posterior rotation. Some patients exhibit entropion of the lower eyelid brachycepthaly, prominent forehead and/or metopic suture, open fontanelles, widow’s peak, and a low set posterior scalp line (Fig 4). Cleft lip/palate, bifid uvula or tongue, shortened lingual frenulum and micrognathia have been described.
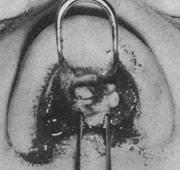
Fig. 11. – Intraoperative photograph showing homograft nasal support (base view)
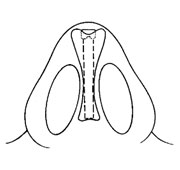
Fig. 12. – Arti’s sketch (base view)
Although the nasal dorsum has been described as elevated or flattened with anteverted nostrils and a flat, inapparent philtrum, review of the literature did not reveal previous reports of agenesis of nasal cartilage in this or any other syndrome. Reports of this syndrome, however, have documented involvement of the upper aerodigestive tract, which can manifest shortly after birth and range from a hoarse cry and coughing to stridor to cyanosis. Cinefluoroscopic studies have shown up to half of each bolus being aspirated, presumably secondary to neuromuscularly dysfunctional deglutition. Hypoplasia of the vocal cords, epiglottis or entire larynx has been reported, and one case displayed a single cavity extending from the epiglottis and hypopharynx superiorly to the trachea and esophagus inferiorly. Review of literature did not reveal any reports of reconstructive rhinoplasty for agenesis of nasal cartilage. It has been hypothesized that these findings, now including agenesis of nasal cartilage, are an example of a developmental field defect.
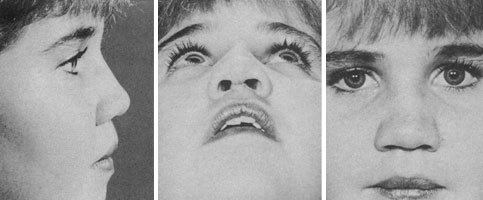
Fig. 13. – Postoperative results.
This case again demonstrates several advantages of comprehensive medical care rather than the limited benefit of efforts focused only on reconstructive technique. First, an understanding of the disease can predict local tissue abnormalities; in this case, anticipating the lack of nasal cartilage allowed us to plan for and use a composite auricular graft and irradiated homograft cartilage. Second, the surgeon can predict systemic manifestations that have implications for the planned surgery; in this case, laryngotracheal compromise influenced our selection of local anesthesia. Finally, the patient can anticipate implication of the disease that extend far beyond the present aesthetic concern: in this case, our patient was counseled regarding genetic transmission and manifestations in her potential off spring.
References
- Optiz JM, Smith DW, Summitt RL. Hypertelorism and hypospadias: a newly recognized hereditary malformation syndrome. J Pediatr. 1965;67:968.
- Optiz JM, Frias JL, Gutenberger JE, Pellett JR. The G syndrome of multiple congenital anomalies. Birth Defects. 1969;5:95-101.
- Optiz JM, Summitt RL, Smith DW. The BBB syndrome familial telecanthus with associated congenital anomalies. Birth Defects. 1969;5:86-94.
- Coté GB, Katsantoni A, Papadakou-Lagóyanni S, et al. The G síndrome of dysphagia, ocular hypertelorism and hypospadias. Clin Genet. 1981;19:473-478.
- Little JR, Optiz JM. The G syndrome, AJDC. 1971;121:505-507.
- Gonzales CH, Hermann J, Optiz JM. Studies of malformation syndromes of man VB: the hypertelorism-hypopasdias (BBB) syndrome. Eur J Pediatr. 1977;125:1-13.
- Optiz JM. G syndrome (hypertelorism with esophageal abnormality and hypospadias or hypospadias-dysphagia, ‘Optiz-Frias’ or ‘Optiz G’ syndrome); perspective in 1987 and bibliography. Am J Med Genet. 1978;28:275-285.
- Cordero JF, Holme LB. Phenotypic overlap of the BBB and G syndromes. Am J Med Genet. 1978;2:145-152.
- Gorlin RJ. Syndromes of the Head and Neck. 3rd ed. New York, NY: Oxford University Press Inc. In press.
- Christian JC, Bixler D. Blythe SC, Merritt AD. Familial telecanthus with associated congenital anomalies. Birth Defects. 1969;5:82-85.
- Coburn TP. G syndrome. AJDC. 1970;120:466.
- Frias Jl. Rosenbloom AL. Two familial cases of the G syndrome. Birth Defects. 1975;11:54-57.
- Arya S, Visekul EF, Gilbert EF. The G syndrome: additional observations. Am J Med Genet. 1980;5:321-324.
- Farndon PA, Donnai D. Male-to-male transmission of the G syndrome. Clin Genet. 1983;24:446-448.
- Cappa M, Borrelli P, Marini R, Neri G. The Optiz syndrome; a new designation for the clinically indistinguishable BBB and G syndromes. Am J Med Genet. 1987;28:303-309.
- Miller PR, Bernstein RM, Pathak A, Decanq HG Jr. Hyperteleorism-hypospadias syndrome with a laryngotracheo-esophageal cleft. J Pediatr. 1977;90:157-158.
©2012 Hilger Facial Plastic Surgery