
Patrick J. Byrne, MD, and Peter A. Hilger, MD
Congenital nasal anomalies are rare Choanal atresia occurs in approximately one in 8000 live births.38 The cleft lip deformity, with its associated nasal deformity, occurs in roughly one in every 1000 live births.5 Congenital nasal masses are estimated to occur in one in every 20,000 to 40,000 live births. The most common of these are nasal dermoids, gliomas, and encephaloceles. These share a common pathogenesis caused by the embryologic development of the nose and anterior cranial base.
EMBRYOLOGY
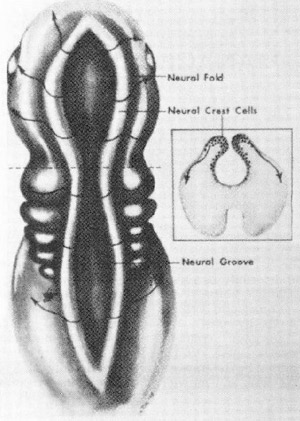
FlG. 1. Dorsal view of a 3 1/2-week-old ombryo. representing neural fold. Inset: Cross-section locating neural crest cells within folds. (Courtesy of Hengerer AS, Oas R: Congenital Anomalies of the Nose: Their Embryology. Diagnosis, and Management. Washington, DC. The American Academy of Otolaryngology, Head and Neck Surgery Foundation, 1987, p 12.)
A basic review of embryology is critical to understanding the pathogenesis of nasal dermoids, gliomas, and encephaloceles. Normal development of the anterior cranial base and nose is dependent on a couple of key processes. One is the formation of the neural tube from ectoderm; the other is the migration of neural crest cells into the mesenchyme to form the skull base and facial structures.38
A midline neural groove develops along the dorsal surface of the embryo by the third week of gestation. This is ectoderm, which as it thickens, deepens, and closes, eventually progresses to form the neural lube, the progenitor of the nervous system. For example, the cephalic end of this tube expands to become the brain. The closure of this neural tube begins in the mid portion of the embryo and progresses anteriorly and posteriorly. Thus, the last areas to close are the most distal (i.e., the anterior and posterior neuropores) (Figs. 1 and 2).
Meanwhile, neural crest cells, originating from the lateral portion of the neural tube, migrate into the mesenchyme between the neural tube and surface ectoderm. This mesenchyme will eventually condense to form the bony and cartilagenous structures of the face. The anterior neuropore area, however, has no neural crest cells, which must migrate from more proximal regions of the tube to reach the area. For this reason, along with the fact that it is the last area of the neural tube to close, the anterior neuropore is particularly susceptible to developmental errors.33
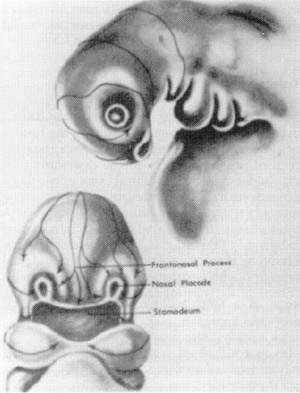
FlG. 2. Migration patterns of neural crest cells. Arrows indicate routes followed by those colls to reach tacial pro¬cesses. (Courtesy of Hengerer AS, Oas R: Congenital Anomalies of the Nose: Their Embryology, Diagnosis, and Management. Washington, DC, The American Academy of Otolaryngology, Head and Neck Surgery Foundation, 1987. p 15.)
The mesenchyme condenses to form mesoderm at various centers, which fuse together and ossify. Before closure, however, there are several potential spaces in the region of the eventual nose and anterior cranial base. Failure of fusion may lead to developmental anomalies. Three adjacent areas in particular have been implicated in the pathogenesis of congenital nasal masses such as dermoids, gliomas, and encephaloceles. These are (1) the fonliculus frontalis, the space between the frontal and nasal bones; (2) the prenasal space, between the nasal bones and nasal capsule, which Ixcomes the nasal cartilages and septum; and 3) the foramen cecum, between the frontal and ethmoid bones (Fig. 3). The crucial characteristic of these areas early in fetal development is that the surface ectoderm and neuroectoderm are in close contact before the proliferation of intervening mesenchyme.
NASAL DERMOIDS
In 1817, Cruvier first described a patient with a midline nasal pit with a small hair growing from it.33 Many terms have been used to describe these lesions, including dermal cyst, dermoid, dermoid cyst, and dermoid sinus cyst. In 1982, Sessions introduced the term nasal dermoid sinus cyst (NDSC) to refer to such lesions with a sinus tract, whereas nasal dermoid cysts do not. NDSCs contain ectoderm (stratified squamous epithelium) and mesoderm (adnexal structures, such as sebaceous glands and sweat glands). Epidermal inclusion cysts, on the other hand, contain skin only. Dermoids also can occur at other sites, including the forehead, anterior fontanelle, orbit and periorbital areas, tongue, and neck. NDSCs account for 3.7% to 12.6% of head and neck dermoid lesions.38 Multiple hypotheses have been offered to account for these lesions. The most popular theory is that the anterior neuropore fails to close properly in the region of the fonticulus frontalis or foramen cecum. Thus, ectoderm can become trapped outside the closing neuropore, and if it remains adherent to the dura as it retracts into the cranium, it can result in a sinus tract connecting the NDSC to the intracranial space17(Fig. 4). Grumwald, who in 1910 implicated inadequate obliteration of the prenasal space of its dural connection32, put another forth. This too involves the abnormal closure of the foramen cecum with epithelium trapped anywhere along the tract, forming the cyst or sinus tract. Another theory says that the middle laver of the fetal trilaminar nasal septum is composed of dura, which normally obliterates. Failure to do so results in ND2C.22 Other theories include aberrant development of skin appendages,16 inclusions in facial clefts, and simple inclusion cysts.16,22
CLINICAL PRESENTATION
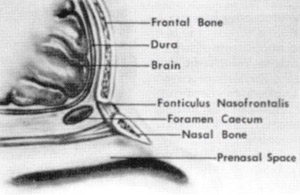
FlG. 3. Anatomy of the base of the anterior cranial fossa and prenasal space during embryologic development. (Courtesy ol Hengerer AS. Oas B: Congenital Anomalies of the Nose: Their Embryology, Diagnosis, and Management. Washington, DC. The American Academy of Otolaryngology, Head and Neck Surgery Foundation, 1987, p45.)
Nasal dermoids typically present at birth or shortly thereafter. Patients present with a nasal mass, a pit, or a fistulous tract. Hair or sebaceous material may protrude from the pit, and some patients present with recurrent drainage.
Most infections are limited to the sinus and cyst, but periorbital cellulitis and osteomyelitis have been reported.28 These occur in the midline, anywhere from the columella to the glabella. Unlike encephaloceles, these masses are solid, do not enlarge with crying, and do not transilluminate. Although not occurring in any known syndromes, a significant percentage of these children may have associated anomalies.33
Most dermoids terminate in a subcutaneous tract, but penetration deep to the nasal bones occurs in up to 45% of patients, with intracranial extension noted in 25% to 30% of cases.13
It is not possible to determine by the physical examination the depth of penetration.
DIAGNOSIS
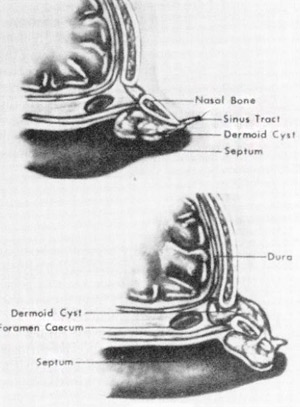
FlG. 4. Top, Localion of dermoid cyst extending deep to the nasal bones. Bottom, Other pathways possible for dermoid cyst and sinus tract development. (Courtesy of Hengerer AS, Oas R: Congenita) Anomalies of the Nose: Their Embryology, Diagnosis, and Management. Washing¬ton, DC, The American Academy of Otolaryngology, Head and Neck Surgery Foundation, 1987: p 46.)
Because of the inherent risk of possible intracranial connection, these lesions should never be biopsied initially. Radiographic examination is necessary to assess the depth of involvement and for the presence of extension into the intracranial space. CT scans demonstrate associated bony anomalies well and may show the classic findings occasionally seen on plain films of swelling or bifidity of the bony nasal septum or widening of the nasal vault. A patent foramen ovale or bifid crista galli are suggestive of an intracranial mass. MR imaging is the imaging study of choice to determine the depth of the tract and is superior to CT in determining intracranial involvement. Intraosseous dermoids may be difficult to visualize, however, and normal fatty deposits associated with development of the frontal sinuses and nasal bones can be read as false positives.2
TREATMENT
Surgical excision of the entire cyst and tract is the treatment. When considering the timing of surgery, the surgeon must balance the concern of possible effects on the developing facial skeleton versus potential bony or cartilaginous destruction, repeated infections, and the ongoing risk of meningitis for those cases with intracranial involvement.23 If an intracranial extension of the mass is known to exist preoperatively, then craniotomy versus craniofacial resection with neurosurgical consultation with complete excision of the lesion is warranted. The approach may be extradural or intradural, and meticulous attention to complete removal is necessary to prevent recurrence. Microsurgical techniques, including the use of the operating microscope and high-speed drills, are helpful for this.18 The extracranial component may be removed at the same time or at a later date. For those lesions known to have no intracranial cyst, the entire tract from the skin to the skull base, if necessary must be removed. Approaches include direct excision with ellipse, lateral rhinotomy, bicoronal flap, and external rhinoplasty.25 Occasionally, the depth of penetration is not clear on the preoperative imaging studies. In such a case, an external approach, with neurosurgical standby, is performed. The surgeon may encounter a stalk extending to the skull base and must determine if this is a simple fibrous band or an epithelium lined stalk. Such an epithelium lined stalk usually indicates the need for an intracranial excision.32 Secondary rhinoplasty is occasionally necessary to repair the resultant cosmetic defect.35
GLIOMAS
Nasal gliomas are congenital developmental anomalies in which glial tissue is situated outside the central nervous system without cerebrospinal fluid connection. They may, however, retain attachments to the dura in 15% to 20% of cases.1” Schmidt coined the term glioma in 1900; this is a misnomer, because the lesions have no neoplastic
potential. They consist of glial tissue, predominantly astrocytes, in a connective tissue matrix. They are classified as extranasal (60%), intranasal (30%), or combined (10%). Intranasal gliomas are more likely to retain attachments to the dura.14 Several theories exist to explain gliomas. Some believe that blastomatous cells destined to become glial tissue simply become trapped outside the central nervous sytem.35 Others believe they result from faulty closure of the anterior neuropore. This prevents the normal inflow of mesoderm that would occur if the neuroectoderm had become separated from the skin as it normally would. A third theory lays the blame with failure of the mesoderm itself to flow into the region of the nose and anterior cranial base. These last two theories support the idea that gliomas and oncephaloceles are developmentally related, with gliomas representing pinched off encephaloceles.27
CLINICAL PRESENTATION
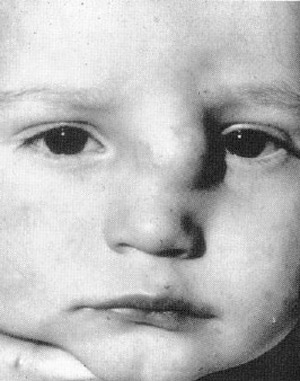
FlG. 5. Extranasal glioma. (Courtesy of R. J. Gorlin. DDS. MS. DSc. Minneapolis. MN )
Gliomas usually present at birth, but have patented as late as the seventh decade of life.12 Unlike dermoids, they usually occur lateral to the midline, as an intranasal mass, an extranasal mass, or both. They are firm, noncompressible, do not expand with crying, and do not transilluminate (Fig. 5) Simultaneous bilateral compression of the jugular veins does not lead to distension of the mass (Furstenburg’s test).
Extranasal masses may exhibit telangiectatic skin overlying them, which may lead the clinician to suspect hemangioma. Intranasally they may prolapse into the nares and be confused with a polyp. They may be attached to the middle turbinate, cribriform plate, or nasal septum. Large gliomas may splay the nasal bones and can cause hypertelorism.
DIAGNOSIS
The diagnostic work-up and Important considerations remain the same. Biopsy is to be avoided. CT and MR imaging are necessary to assess the depth of involvement. Gliomas appear as mixed intensity masses, which on CT may appear continuous with intracranial structures.” Contrast may flow into an encephalocele, but will not in a glioma, allowing the distinction between these two entities.
TREATMENT
Surgical excision is the treatment. The surgeon should be prepared to make the final confirmation of the diagnosis at the time of the operation, despite the results of preoperative imaging External gliomas may be excised by lateral rhinotomy or the external rhinoplasty approach. Intranasal gliomas with large dural attachments likely will need a craniotomy with anterior fossa exploration. If the diagnosis is in doubt, the procedure can by entirely extracranial until it is determined if an encephalocele is in fact present or if a large dural defect exists. Intranasal endoscopic surgery may be used to aid in this determination and has been used to completely excise intranasal gliomas without intracranial extension.37
ENCEPHALOCELES
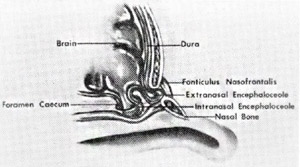
FlG. 6. Pathways taken by neural elements that load to formation of internal or uxlornal encephaloceles (Courtesy ol Hengerer AS, Oas R: Congenital Anomalies ol the Nose: Their Embryology. Diagnosis, and Management. Washington. DC. The American Academy of Otolaryngology. Head and Neck Surgery Foundation. 1987, p68.)
Encephaloceles are herniations of neural tissue through a bony delect. They .ire more common along the spine, but 15% to 25% occur in the anterior frontal area.38 The theories of pathogenesis are the same as for gliomas. Encephaloceles may be classified according to the contents of the herniated sac. Each contains leptomeninges and dura. The presence of meninges distinguishes these lesions from gliomas. Those containing only these structures are referred to as meningoceles. If brain is also present, it is termed encephalomeningocele. Encephaloceles with a connection to the ventricular system within the sac are encephalomeningocystoceles (Fig. 6).
A more useful classification is by site, which determines the clinical symptoms. They are classified as either occipital, sincipital, or basal. The occipital form occurs in the occiput. The other two forms occur in or about the nose. The sincipital is the more common of the two, and is further subdivided into three subtypes. The nasofrontal subtype exits the skull between the nasal and frontal bones. These deform the orbit by pushing the medial orbital walls laterally. The nasoethmoidal subtype exits between the nasal bones .mil nasal cartilages, thus leaving the connection of the nasal and frontal bones intact. The naso-orbital subtype is less common than the first two and passes through a defect in the frontal process of the maxilla. Basal encephaloceles are less common than the sincipital and are subdivided into tour subtypes. The transethmoidal and sphenoethmoidal pass through the cribriform plate. Sphenorbital encephaloceles are rare, passing through the superior orbital fissure, producing exopthalmos. Transsphenoidal encephaloceles pass through a defect posterior to the cribriform plate into the nasopharynx.
CLINICAL PRESENTATION

FlG. 7. Encephalocele. (Courtesy of R. J. Gorlim. DDS. US. DSc. Minneapolis, MN.)
Sincipital encephaloceles present as soft, compressible masses over the glabella. They are often pulsatile and expand with crying or with the Furstenburg test. They may, however, have none of these classic signs. The basal types are typically not visible externally, although they may cause hypertelorism or widening of the root of the nose. Nasal obstruction is the most common presenting symptom. Like gliomas, they can present as a nasal polyp. They are, however. located medial to the middle turbinate, as opposed to gliomas, which are attached laterally (Fig. 7).
DIAGNOSIS
CT scans and MR images are the studies of choice. No intranasal masses in the infant should be dismissed as a polyp. Bony dehiscences of the cranium may be seen and are considered an encephalocele until proven otherwise. MR images can demonstrate the intracranial extension of the encephalocele.2 Biopsy or aspiration is contraindicated.
TREATMENT
Encephaloceles expand with time, and this has been shown to cause increasing hydrocephalus and increased herniation of brain tissue into the sae.32 For this reason, along with the persistent risk of meningitis, most surgeons operate early, usually within the first few mouths of life.17, 33 A joint procedure with the neurosurgeon is indicated for these lesions. Sincipital encephaloceles may be resectable by an extracranial approach, but this is unusual.38 A craniotomy is typically required. The extracranial component may be removed at the Bame lime, although some have recommended observation because they may atrophy.8 The hypertelorism is said to resolve in young children,37 whereas adults with a cosmetic deformity will likely require corrective surgery.
CHOANAL ATRESIA
Roederer first described unilateral or bilateral obstruction of the posterior choana of the nose in 1755.14 The female to male ratio is 2:1, whereas unilateral atresia is twice as common as bilateral atresia. The atresia plate is bony 90% of the time and membranous 10%. Associated anomalies occur in 50%. The CHARGE association is the most significant of these, consisting of coloboma of the retina, cardiac defects, mental retardation, genitourinary anomalies, and hearing loss in addition to choanal atresia.
EMBRYOLOGY
The simplest theory to explain choanal atresia is persistence of the nasobuccal membrane. Several others have been proposed, including overgrowth of palatine bone and persistence of the buccopharyngeal membrane, a theory no longer recognized. Misdirection of mesodermal flow of the neural crest cells in the region secondary to local factors have been Implicated in persistence of the nasobuccal membrane.14
DIAGNOSIS
Cyclical respiratory distress and cyanosis, relieved by crying, is the hallmark of bilateral choanal atresia. Infants are obligate nasal breathers tor the first 4 to 6 weeks of life, thus making bilateral choanal atresia a potentially fatal emergency. A quick way to diagnose the problem is by attempting to pass a 6 or 8 Fr catheter through the nose. If it cannot be passed into the oropharynx, choanal atresia is likely. Likewise, a mirror held in front of the nostril will fail to fog in the presence of choanal atresia. CT scanning will make the diagnosis.
Unilateral atresia usually presents later in life. Unilateral nasal obstruction and chronic rhinorrhea are the cardinal signs and may not be evident until several years later Obstruction of the uninvolved side may precipitate airway obstruction. An association between unilateral and bilateral choanal atresia with sudden infant death syndrome has been suspected.38
TREATMENT
Emergency treatment consists of establishing an oral airway, which breaks the seal of the tongue and epiglottis against the palate. A tracheotomy is rarely needed. Several techniques for definitive correction have been established. The earliest technique is transnasal, first developed as a blind puncture by Emmert in 1853.15 Serious complications have been reported, including passage of the trocar into the brainstem. The transnasal approach now uses the CO2 laser. These techniques have gained in popularity in recent years, with good results reported.15 The transnasal approach was generally believed to have a higher incidence of restenosis than the transpalatal approach.14
The transpalatal approach has traditionally been the most favored. The posterior end of the hard palate is removed followed by the removal of the posterior vomer. The transseptal approach involves making a window in the septum anterior to a unilateral atretic plate.
I he transseptal approach is for unilateral atresia, in which a window is created in the posterior septum, allowing passage of air into the nonatretic side.
CLEFT LIP NASAL DEFORMITY
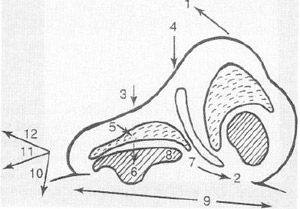
FlG. 8. Components of the unilateral cleft lip nasal deformity. 1 = Nasal deviation to the affected side; 2 = Septal deviation to the normal side. 3 = Lowered dome; 4 = Bifid tip; 5 = Distorted crura. The size of the lateral crus is normal, but the position and shape are abnormal. It extends as a bridge across the cleft. The cephalic lateral crus is rolled in (inward buckling of the ala). The medial crus is inferiorly displaced. The angle between the medial and lateral crus is obtuse: 6 = Downward movement of the edge of the cartilage and nostril: 7 = Shortened columella of the cleft side. 8 = Acute nostril angle and naris circumference greater than that of the normal side; 9 = Widened alar base; 10 = Caudally displaced alar base; 11 = Posteriorly displaced alar base; 12 = Laterally displaced alar base.
Cleft lip or palate occurs in approximately 1 in 680 births. The cause is considered multifactorial, with numerous conditions contributing to their development. Of the patients with these deformities, 20% to 30% have an isolated cleft lip, 35% to 57% have cleft lip and palate, and 30% to 45% have isolated cleft palate.5
Most patients with cleft lip have an associated nasal deformity, with varying degrees of severity. The appearance of this nasal deformity tends to become more apparent with increasing age, in contrast to the repaired lip, which seems to improve with age.5 The nasal deformities frequently associated with the cleft lip have been well described by Cutting et al9 (Fig. 8). The columella is shortened on the cleft side, with the columellar base directed into the noncleft side. The anterior nasal spine is deviated into the noncleft side, and the nasal lip is deviated toward the cleft. The medial crus tends to slump laterally, while hooding of the lower lateral cartilage and alar rim occurs caudally
(Fig. 9). There is insufficiency of vestibular skin and hypoplasia of the maxilla on the involved side.23
Approximately 60% of patients with cleft lip or palate have an airway that is inadequate for normal nasal breathing.36 The difference in airway size between the cleft and noncleft nose is approximately 30%. The type of cleft seems to affect the nasal airway size, at least in children.36 The airway is smaller with unilateral cleft lip and palate, but all clefts tend to compromise the nasal airway to some extent. The incidence of mouth breathing, which raises concerns about effects on nasal and midfacial growth, is higher in the deft population.36
Development of an adequate airway, therefore, is a goal of cleft lip rhinoplasty. The other goals may include lengthening of the columella on the cleft side, correction of the deformed ala, repositioning of the alar base, creating tip symmetry, and reconstructing the nasal floor and sill.5 Primary nasal repair at the time of primary lip repair has gained greater acceptance in recent limes. Past concerns regarding the effects of surgery on disrupting growth centers, damaging the delicate cartilages of the infant’s nose, or causing scarring that will compromise future efforts at definitive repair have been tempered. Long-term follow-up has demonstrated excellent symmetry and maintenance of nasal form into young adulthood with primary rhinoplasty techniques. This is accomplished before the age of recall, thus sparing the child from much of the ridicule from classmates in school. In addition, it takes advantage of the pliability of the infantile nasal cartilages, allowing excellent results from relatively conservative techniques.26
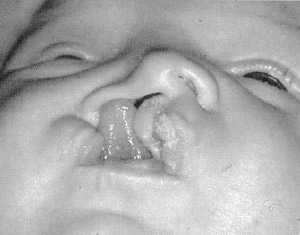
FlG. 9. Cleft lip nasal deformity. (Courtesy of J. D. Sidman. MD. Minneapolis, MN.)
Many techniques for primary repair of the cleft lip nasal deformity have been described.5, 24, 26 At the time of primary lip repair, several objectives may be addressed to correct the nasal deformity. The base of the ala is placed in a more symmetric position in relation to the noncleft side and may as well be cinched to present a more normal appearance. The columella may be straightened and lengthened. The skin overlying the medial and lateral crura on the cleft side is widely undermined to allow repositioning of the lateral crus. Bolsters may be used to aid in maintaining the new position of the cartilage, thus providing improvement of the assymetries of the nasal tip and alar cartilages.5
Secondary rhinoplasty refers to the intermediate and the definitive repairs. The intermediate procedure to reconstruct the nasal tip is often necessary when the patient is between 4 and 12 years of age. Earlier repair allows the child to avoid some of the psychosocial pressures from other children in school. Waiting until between 8 and 12 years of age, after orthodontic alignment and alveolar bone grafting, provides a better skeletal base for correction of severe nasal deformities. The procedure may be performed in conjunction with revision of the lip scar. The definitive rhinoplasty is performed when maxillary and nasal growth has been completed, generally between 16 and 18 years of age. More aggressive septoplasties, osteotomies, and grafting may be performed at this lime. Open rhinoplasty techniques have been helpful in gaining more control of the correction in these difficult operations. Individualization of the approach to the patient’s particular needs is mandatory for optimal results.
HEMANGIOMA
Hemangiomas are the most common benign tumor affecting infants and children. They occur throughout the head and neck, including the nose. Hemangiomas of the nose may be internal or external. They typically present at birth or in the first few months of life. A rapid proliferative phase lasting 6 to 12 months is usually followed by spontaneous involution. Because of this, conservative management is usually appropriate.
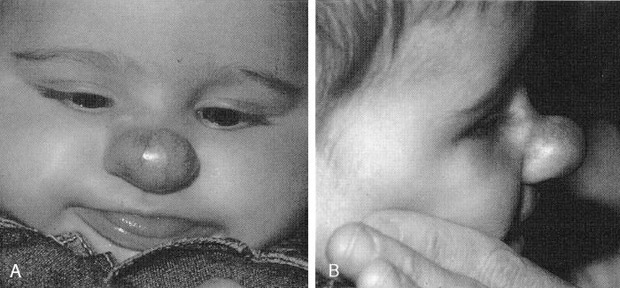
FlG. 10. A and B, Nasal tip hemangioma producing Cyrano de Bergoroc deformity.
On occasion, certain characteristics lead one to treat hemangiomas. Involution may fail to occur. Lesions of the nasal lip may produce a classic “Cyrano de Bergerac” deformity, which is accompanied by a significant amount of ridicule by other children (Figs. 10A and B). Nasal obstruction may occur. The delicate cartilages of the growing nose may undergo aplasia secondary to mass effect.29 In addition, residual contour irregularities may persist secondary to scarring, even after involution or excision.
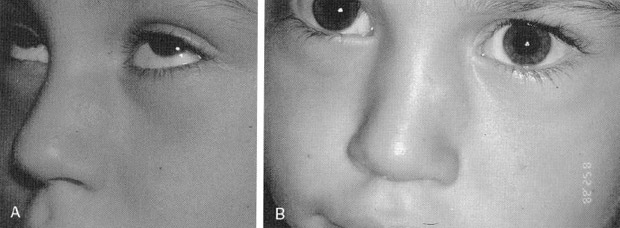
FlG. 11. A, Nasal hemangioma pretreatment. B. Posttreatment with pulse dye laser. (Courtesy of J. D Sidman. MD. Minneapolis. MN.)
Several treatment options exist to address this difficult problem. Intralesional injection of sclerosing agents or corticosteroids may be effective. The KTP, CO2 argon, and Nd: YAG lasers have been used successfully to remove hemangiomas30 (Figs. 11A and B). Lasers may by used alone or in conjunction with traditional surgical techniques. Embolization of nasal hemangiomas has been reported.7, 34 Surgical treatments focus on adequate exposure to ensure complete removal of tumor and to address the underlying cartilaginous abnormalities. As with other indications for pediatric nasal surgery, conservative surgery must be performed in the nasal growth centers, including the soptovomerine suture, nasal bones, and vestibular skin.
ARHYNIA
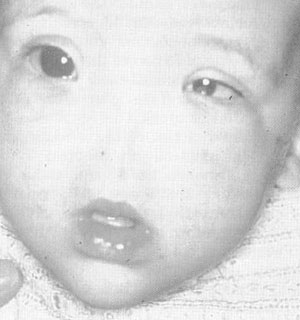
FlG. 12. Arhynia. (Courtesy of R. J. Gorlin. DDS. MS. DSc, Minneapolis. MN.)
Complete agenesis of the nose is rare. The external nose, nasal cavities, and the olfactory system are absent. The diagnosis is obvious at birth (Fig. 12). The anomaly may occur alone, but more often it is associated with other congenital anomalies, particularly of the eye and central nervous system. These may be serious enough to be incompatible with life. When respiratory distress is present, these patients are to be managed as a child with choanal atresia would be managed. Several authors have noted a rapid progression to mouth breathing and minimal swallowing difficulties.15, 38
Treatment may be delayed because most patients breathe adequately. The initial treatment is to create a nasal passage. This is performed by drilling away the bone in the area and lining the created passageway with skin grafts. The definitive repair requires a multistage total nasal reconstruction with a forehead flap and cartilage or bone grafting. This may be delayed until facial growth is complete.
POLYRHINIA
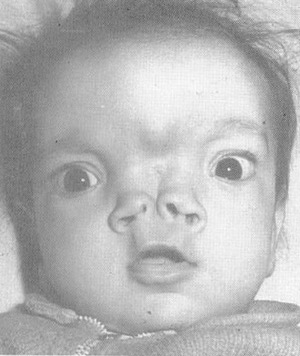
FlG. 13. Polyrhinia. (Courtesy of R. J. Gorlin. DDS, MS, DSc, Minneapolis, MN.)
Double nose deformity, or polyrhinia, is extremely rare. It is distinguished from median nasal cleft, in which a single nose is divided into two, each with a single nostril. In polyrhinia, two completely formed noses are present (Fig. 13). A duplication of the medial nasal processes during embryonic development is believed to account for this entity.38 Treatment consists of excision of the medial halves of each nose. A coexistent choanal atresia may be present and need to be dealt with.38
NASAL CLEFTING
Clefting of the nose is rare. Although it may occur alone, it is more often associated with other abnormalities, such as clefts of the lip and palate, ocular hypertelorism, and anterior cranium bifidum occultum. These deformities may belter be categorized under the broad term frontonasal dysplasia10(Fig. 14).
Median nasal clefts range from a simple scar at the cephalic end of the nasal dorsum to a nose completely split into two halves (Fig. 15). The airway is usually not obstructed. Severe cases will require an evaluation to rule out encephalocele prior to reconstruction.14
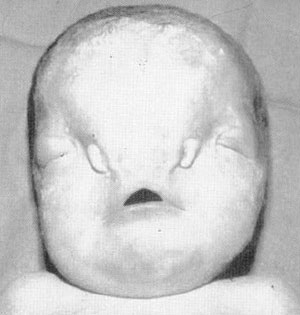
FlG. 14. Frontonasal dysplasia. (Courtesy of R. J. Gorlin, DDS. MS. DSc. Minneapolis, MN.)
Lateral nasal clefts also show great variability. They may range from a small groove of the lateral aspect of the nasal ala (Fig. 16), to a large furrow extending all the way to the medial canthus. The cause or the lateral cleft involves failure of mesenchymal flow between the medial and lateral nasal processes.14 The nasolacrimal duct is often involved and may need to be addressed at the time of reconstruction. CT scanning is helpful in determining the bony anatomy, and MR imaging may evaluate for the presence of encephalocele. It is advisable to delay reconstruction to allow as much normal nasal development to occur as possible. The repair typically involves local rotation flaps with composite auricular skin and cartilage grafts.
PROBOSCIS LATERALIS
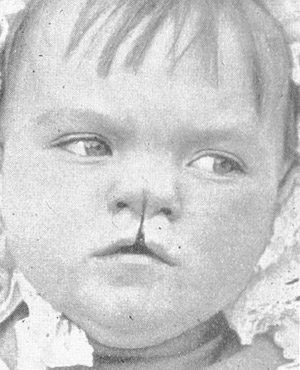
FlG. 15. Median nasal cleft of lip and nose. (Courtesy of R. J. Gorlin, DDS, MS, DSc. Minneapolis, MN.)
Sclenkoff first described proboscis lateralis, or congenital tubular nose, in 1884.38 It remains a rare anomaly. During embryological development, the medial and lateral nasal processes, as well as the globular process, fail to develop on the involved side. The maxillary process on the involved side, therefore, fuses with the opposing globular and nasal processes.14 Along this line of fusion of the maxillary process and the contralateral nasal process forms a tubular skin and soft tissue structure attached to the medial can thus. The result is a tubular structure attached to the medial canthus (Pig. 17). Histologically, the proboscis contains sebaceous, sweat, lacrimal gland tissue, cartilage, and hair.38 There may be a normal nasal cavity or complete lack of nasal cavity and paranasal sinus format ion on the involved side.
The nasolacrimal duct forms from buried ectoderm at the line of fusion of the medial and lateral nasal processes. This structure, therefore, may need to be rerouted at the time of reconstruction. As much of the soft tissue of the proboscis as is necessary to reconstruct is saved; the rest of the lesion is excised. An associated skeletal deficiency may be corrected by the use of bone or cartilage grafts.
CONGENITAL NASAL PYRIFORM APERTURE STENOSIS
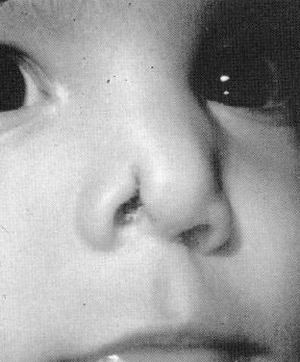
FlG. 16. Lateral nasal cleft (Courtesy of R. J. Gorlin, DDS. MS, DSo.)
Congenital nasal pyriform aperture stenosis (CNPAS) was first described as a distinct entity in the radiology literature in 1988 and clinically in 1989.1, 4 It has received attention as a cause of pediatric airway obstruction in the past decade. The pyriform aperture is the
narrowest section of the nasal airway. Because the cross-sectional area is related to the fourth power of the radius, very small changes in diameter of the newborn aperture may produce significant obstruction. Although the embryology of this condition is unknown, overgrowth of ossification of the maxillary prominence has been postulated.20
Other anomalies have been associated with CNPAS.1 CNPAS as a manifestation of holoprosencephaly has been suggested because a single central incisor has been noted with CNPAS, along with pituitary hypofunction1 (Fig. 18). Most reported cases, however, have been isolated defects.
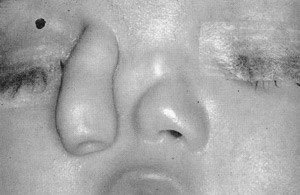
FlG. 17. Proboscis lateralis.(Courtesy of R.J. Gorlin, DDS. MS. DSc. Minneapolis, MN)
The patient with CNPAS presents with cyclical cyanosis relieved by crying, similar to choanal atresia. A bony shelf maybe noted that prevents passage of a fiberoptic scope. Tin-diagnosis is confirmed by CT scanning. The typical findings include a narrow nasal inlet, triangle shaped palate, bone overgrowth of the nasal process of the maxilla, and abnormal dentition. The width of the pyriform aperture may be measured, and a width less than 11 mm in a term infant has been suggested as diagnostic.
Initial management involves establishing an airway with a McGovern nipple or a nasal stent. Unlike choanal atresia, however, the management may not require surgery.20 Patients with milder degrees of stenosis may be observed because they may outgrow the stenosis. These patients may be treated with humidification and topical decongestants. The parents must be educated about the risk of airway obstruction in the infant with noncritical CNPAS who develops an upper
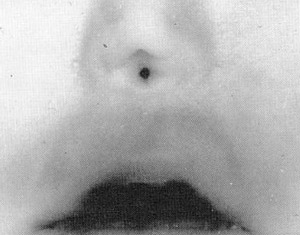
FlG. 18. Congenital nasal pyriform aperture stenosis in patient with holoprosencephaly (Courtesy of R. J. Gorlin, DDS. MS, DSc.)
respiratory tract infection. In more severe cases, surgical intervention is required If possible, waiting 6 to 8 weeks for nasal growth has been advised.20 A sublabial approach to the pyriform aperture may then be performed. Resection of the bony obstruction is achieved by drilling the pyriform aperture back to the nasolacrimal duct and head of the inferior turbinate. Stents are placed bilaterally and left in place for 1 to 4 weeks.31
SUMMARY
Congenital nasal anomalies are rare disorders that may present challenges to the facial plastic and reconstructive surgeon. These anomalies are extremely varied and result from a wide range of disordered embryonic development. A basic understanding of nasal and facial development is helpful in understanding these anomalies. In addition, proper respect for the unique challenges of nasal surgery on infants and children is crucial to best serve the patient. This allows a proper work-up and consideration of appropriate treatment options before surgical repair.
REFERENCES
- Arlis H, Ward R: Congenital nasal pyriform aperture stenosis: Isolated abnormality vs developmental field defect. Arch Otolaryngol Head Neck Sure 118:898-991, 1992
- Barkovich A, Vandermarck P, Edwards M, et al: Congenital nasal masses: CT and MR imaging features in 16 cases. American Journal of Neuroradiology 12:105-116, 1991
- Belden C, Mancuso A, Schmalfuss I: CT features of congenital nasal pyriform aperture stenosis: lnitial experience. Radiology 213:495-501, 1999
- Brown O, Myer C. Manning S: Congenital nasal pyriform aperture stenosis. Laryngoscope 99:86-91, |yH9
- Bumsted R: Cleft lip and palate. In Papel I, Nachlas N (eds): Facial Plastic and Reconstructive Surgery. St. Louis, Mosby-Year Book. 1W, pp 546-556
- Burget G, Menick F: In Aesthetic Reconstruction of the Nose. St. Louis, Mosby-Year Book, 1994
- Burrows P. Lasjaunias P, Ter Brugge K, et al. Urgent and emergent embolization of lesions of the head and neck in children. Pediatrics 80:386-304, 1987
- Choudhury A, Taylor J: Primary Intranasal encephalocele: Report of four cases. J Neurosurg 59:1111, 1963
- Cutting C, Bardach J, Pang R: A comparative study of the skin envelope of the unilateral cleft lip nose subsequent to rotation-advancement and triangular flap lip repairs. Hast Reconstr Surg 84:409-117, 1989
- DeMyer W: The median cleft face syndrome: Differential diagnosis of cranium bifidum occultum, hypertelorism, and median cleft nose, lip, and palate. Neurology 17:961-971, l967
- Ey E, Han B, Towbin R, et al: Bony inlet stenosis as a cause of nasal airway obstruction. Radiology 168:477-479, 1988
- Fitzpatrick E, Miller R: Congenital midline nasal masses: Dermoids, gliomas, and encephaloceles. J La State Med Soc 148:93-96, 1998
- Fornadly J, Tami T: The use of MRI in the diagnosis of nasal dermoid sinus cyst. Otolaryngol Head Neck Surg 101:397-398,1989
- Hengerer A, Oas R: Congenital Anomalies of the Nose: Their Embryology, Diagnosis, and Management, ed 2. American Academy of Otolaryngology-Head and Neck Surgery Foundation, Inc 2:25-83, Alexandria, VA 1987
- Hengerer A, Yanofsky S: Congenital malformations of the nose and paranasal sinuses. In Bluestone C, Stool S, Kenna M (eds): Pediatric Otolaryngology. Philadelphia, WB Saunders, 1996, pp 831-842
- Hoshaw T, Walike J: Dermoid cysts of the nose. Arch Otolaryngol Head Neck Surg 93:487-491,1971
- Hughes G, Sharpino G, I lunt W, et al: Management of the congenital midline nasal mass: A review. Head and Neck Surgery 2:222-233,1980
- Jaffe B: Classification and management of anomalies of the nose. Otolaryngol Clin North Am 14:989-1004, 1981
- Johnson P, Rydlund K, 1 lollins R: Congenital nasal pyri-form aperture stenosis. Plast Reconstr Surg 1696-1699, 1999
- Jones J, Young E, Heier L: Congenital bony nasal cavity deformities. Am J Rhinol 12:81-86,1998
- Kubo K, Garrett W, Musgrave R: Nasal gliomas. Plast Reconstr Surg 52:47-51,1973
- Littlewood A: Congenital Anomalies of the Nose: Their Embryology, Diagnosis, and Management, ed 2. American Academy of Otolaryngology-Head and Neck Surgery Foundation, Inc. 2:25-83, 1987
- McCaffrey T, McDonald T, Gorenstein A: Dermoid cysts of the nose: Review of 21 cases. Otolaryngol Head Neck Surg 87: 52-59,1979
- Madorsky S, Wang T: Unilateral cleft rhinoplasty: A review. Otolaryngol Clin North Am 32:669-682,1999
- Mankarious L, Smith R: External rhinoplasty approach for extirpation and immediate reconstruction of con-genital midline nasal dermoids. Ann Otol Rhinol Laryngol 9:786-789,1998
- Millard D, Morovic C: Primary unilateral cleft nose correction: A 10 year follow-up. Plast Reconstr Surg 102:1331-1338,1998
- Morgan D, Evans J: Developmental nasal anomalies. J Laryngol Otol 104:394-403,1990
- Pratt L: Midline cysts of the nasal dorsum: Embryologic origin and treatment. Laryngoscope 75:968-980, 1965
- Pitanguy I: Surgical treatment of hemangiomas of the nose. Ann Plast Surg 36:586-592,1996
- Ries W, Aly A, Nachlas N: Lasers in facial plastic surgery. In Papel I, Nachlas N (eds): Facial Plastic and Reconstructive Surgery. St. Louis, Mosby-Year Book, 1992, pp 84-98
- Rombaux P, Hamoir M, Francois G, et al: Congenital nasal pyriform aperture stenosis in newborns: Report on three cases. Rhinology 38:39-42, 2000
- Sessions R, Hudkins C: Congenital anomalies of the nose. In Bailey B: Head and Neck Surgery-Otolaryngology, vol 1. Philadelphia, JB Lipincott, 1993
- Sessions R: Nasal dermoid sinuses: New concepts and explanations. Laryngoscope 92 (suppl 29):l-28,1982
- Webb C, Porter G, Spencer M, et al: Cavernous haeman-gioma of the nasal bones: An alternative management option. J Laryngol Otol 114:287-289,2000
- Wardinsky T, Pagon R, Kropp R, et al: Nasal dermoid sinus cysts: Association with intracranial extension and multiple malformations. Cleft Palate Craniofac J 28:87-95,1991
- Warren D, Drake A: Cleft nose: Form and function. Clin Plast Surg 20:769-779,1993
- Yokoyama M, Inouye N, Mizuno F: Endoscopic man-agement of nasal glioma in infancy, hit J Pediatr Otorhi-nolaryngol 51:51-54,1999
- Zeitouni A, Shapiro R: Congenital anomalies of the nose and anterior skull base. In Tenfik T, Derkaloushan: Congenital Anamolies of the Ear, Nose, and Throat. New York, Oxford University Press, 1997